Новая технология картирования белков раскрывает внутреннюю работу клеток
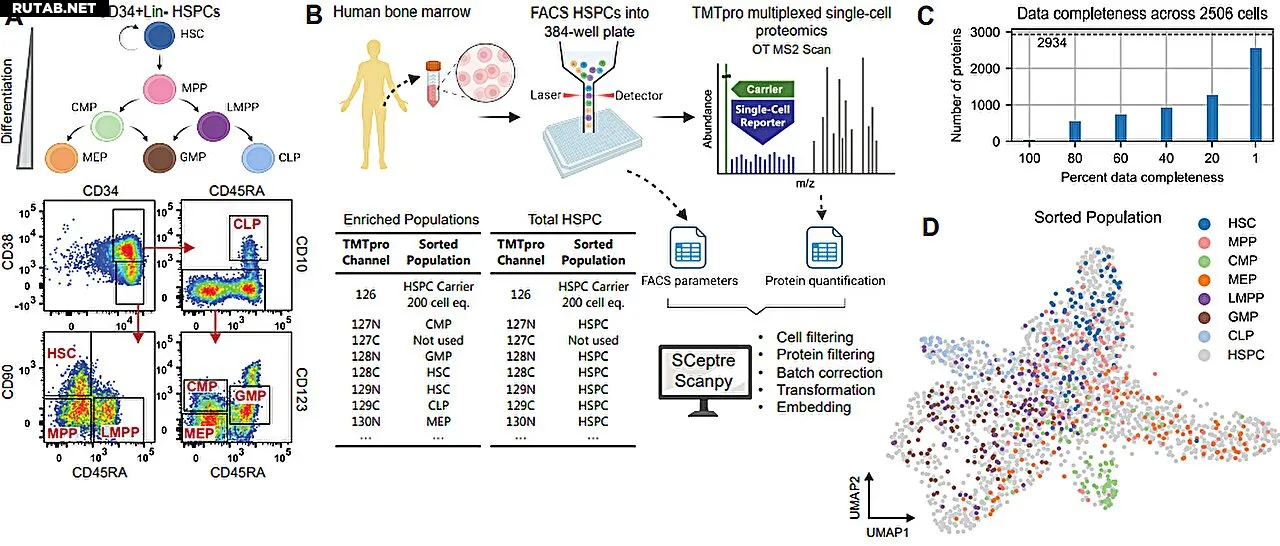
Данные протеомики отдельных клеток FACS-изолированных человеческих HSPC. Автор: Бенджамин Фюртвенглер и др.
За последнее десятилетие значительно возрос интерес к изучению экспрессии нашего генетического кода на уровне отдельных клеток, чтобы определить функции и активность любой клетки в ходе здоровья или болезни.
Идентичность клетки и то, как эта идентичность может нарушиться, критически важны для её роли во многих крупнейших проблемах здравоохранения, включая рак, нейродегенеративные заболевания, а также генетические и нарушения развития. Приближение к отдельным клеткам позволяет нам различать варианты, которые в противном случае были бы потеряны в усреднённых данных по региону. Это необходимо для поиска новых медицинских решений для лечения заболеваний.
Большинство экспериментов по экспрессии генов в отдельных клетках используют технологию под названием секвенирование РНК отдельных клеток (scRNA-seq), которая создаёт карту того, какие именно гены копируются в короткие «транскрипты» внутри ядра. Однако секвенирование РНК отдельных клеток (scRNA-seq) даёт нам лишь окно в промежуточный шаг между генетическим кодом и белками, которые выполняют (почти) все задачи в нашем организме.
Учёные уже некоторое время знают, что уровни мРНК не точно соответствуют уровням их соответствующих белков в клетках. На это могут влиять многие факторы, включая сложные способы, которыми клетки контролируют стабильность мРНК и их трансляцию в белки, а также то, как белки деградируют, всё это контекстно-зависимым образом.
Преодоление этой проблемы
Учёные из Лаборатории Финсена при госпитале Ригсхоспиталет, Центра биотехнологических исследований и инноваций (Копенгагенский университет), Технического университета Дании (DTU) и Центра Гельмгольца в Мюнхене использовали новый подход для анализа сложной популяции белков в отдельных клетках во время формирования клеток крови.
Этот протеомный анализ отдельных клеток означает обход промежуточных звеньев мРНК и построение карты белков, присутствующих в клетках во время их дифференцировки из стволовых клеток в зрелые клетки крови.
Один из старших авторов исследования, Бо Порсе из Лаборатории Финсена при госпитале Ригсхоспиталет и Центра биотехнологических исследований и инноваций (Копенгагенский университет), говорит:
«Процесс клеточной дифференцировки невероятно сложен, и нам нужно полностью понимать нюансы того, что происходит внутри каждой клетки на каждом этапе её жизни, чтобы решать случаи, когда процесс идёт неправильно.
С этим исследованием мы показали возможность использования этой технологии для точного моделирования точных стадий экспрессии генов, охватывающих как синтез и распад мРНК, так и последующий синтез и распад белков на протяжении клеточной дифференцировки».
Это исследование, опубликованное в журнале Science, представляет первое использование технологии, совместно разработанной DTU, госпиталем Ригсхоспиталет и Копенгагенским университетом, а именно протеомики отдельных клеток с помощью масс-спектрометрии (протеомики отдельных клеток с помощью масс-спектрометрии (scp-MS)) в биологически релевантной системе органов, в отличие от лабораторных клеточных культур.
Хотя пока невозможно обнаружить каждый белок, присутствующий в каждой клетке, исследователи смогли сравнить данные мРНК из традиционного (десятилетней давности) метода секвенирования РНК отдельных клеток (scRNA-seq) и этого нового анализа белков отдельных клеток, и обнаружили, что в более дифференцированных клетках крови два набора данных сильно коррелировали (то есть изменения уровней мРНК коррелировали с изменениями уровней их соответствующих белков), однако в стволовых клетках и более незрелых клетках наборы данных плохо коррелировали.
Это предполагает, что оборот транскриптов мРНК, скорость их трансляции или стабильность белков, экспрессируемых в клетках на ранних стадиях их дифференцировки, могут меняться по мере того, как клетки становятся более дифференцированными.
«Это исследование является кульминацией многих лет интенсивной разработки технологий. Не так давно идея измерения тысяч белков в отдельных человеческих стволовых клетках из костного мозга казалась научной фантастикой», — говорит со-старший автор Эрвин Шуф, доцент и руководитель Лаборатории клеточного разнообразия на факультете биотехнологии и биомедицины Технического университета Дании.
«Мы никогда не представляли, что сможем применить протеомики отдельных клеток с помощью масс-спектрометрии (scp-MS) к чему-то столь сложному и динамичному, как человеческая кровеносная система, так скоро. Но вот мы здесь, наконец-то смогли получить доступ к слоям биологии, которые полностью невидимы для методов, основанных только на РНК. Это свидетельство силы масс-спектрометрии, считывания на уровне белков и основанной на данных системной биологии для преобразования нашего понимания того, как клетки принимают решения о судьбе».
Результаты и влияние
Исследователи продолжили изучение некоторых белков, которые, по-видимому, снижались в количестве во время клеточной дифференцировки, несмотря на стабильные уровни мРНК на протяжении всего процесса. Редактируя генетический код для удаления (или «нокаута») этих генов, учёные показали, что это привело к сокращению количества стволовых клеток.
Это предполагает, что эти белки необходимы для поддержания здоровой популяции стволовых клеток в системе, чтобы обеспечить достаточную поставку клеток крови в организме. Просто анализируя данные секвенирования РНК отдельных клеток (scRNA-seq), эти функционально релевантные белки никогда не были бы идентифицированы, и их роли в этом важном процессе остались бы скрытыми.
«Интегрируя измерения РНК и белков в динамическую модель, мы можем охватить полный жизненный цикл экспрессии генов в отдельных клетках. Это помогает нам понять не только то, что написано в генетическом сценарии, но и как это выполняется в реальном времени. Я увлечен тем, как эти измерения белков с разрешением на уровне отдельных клеток всё больше открывают полностью новые окна в клеточную биологию», — говорит Фабиан Тейс, директор Вычислительного центра здоровья в Гельмгольце Мюнхен и профессор математического моделирования биологических систем в Техническом университете Мюнхена.
Эта работа знаменует собой поворотный момент для биологии отдельных клеток: возможность прямо измерять белки с разрешением отдельных клеток в первичной человеческой ткани. Это открывает дверь к открытию скрытых слоёв регуляции в развитии, болезни и регенерации, слоёв, которые одна РНК никогда не могла показать.
Как телескопы преобразуют наше понимание космоса, протеомика отдельных клеток теперь делает то же самое для внутренней работы жизни.
Больше информации: Бенджамин Фюртвенглер и др., Mapping early human blood cell differentiation using single-cell proteomics and transcriptomics, Science (2025). DOI: 10.1126/science.adr8785. www.science.org/doi/10.1126/science.adr8785
Источник: University of Copenhagen






0 комментариев